¿Qué es el síndrome de Ehlers-Danlos?
Resumen del contenido

Pediatra, experta en acupuntura y nutrición oncológica. Actualmente lidera la Unidad de Oncología Pediátrica Integrativa del Hospital Sant Joan de Déu Barcelona. Ha sido pionera en España desarrollando la acupuntura infantil como un procedimiento médico complementario, seguro e indoloro. Compagina su trabajo con la docencia.
Este síndrome es un conjunto de enfermedades heterogéneas hereditarias relacionadas con la alteración genética tanto de la estructura como la síntesis del colágeno. El colágeno es una proteína resistente y fibrosa, que tiene un papel esencial en la unión, la consolidación de las células y proporciona elasticidad a los tejidos corporales.
¿Qué es y a quién afecta?
Es una enfermedad rara que fue descrita por primera vez por Hipócrates en el año 400 aC en su libro Sobre los aires, las aguas y los lugares al observar el aspecto de las rodillas de los nómadas escitas.
El primer estudio completo de este síndrome lo realizó el físico ruso Tschernogbow en 1892. Su publicación no llegó a Europa y las investigaciones continuaron allí de manera paralela. Por eso en Rusia el Síndrome de Ehlers-Danlos se ha conocido desde entonces como Síndrome de Tschernogbow.
En 1901 el dermatólogo danés Edvard Lauritz Ehlers y en 1908 Henri Alexandre Danlos presentaron casos clínicos de este trastorno. No fue hasta 1936 cuando Frederick Parkes-Weber sugirió el nombre de Síndrome de Ehlers-Danlos en honor a los dos médicos anteriores.
La prevalencia se estima en 1 por cada 5.000 o 10.000 personas. Afecta por igual a hombres y mujeres de todas las razas y grupos étnicos.
Actualmente, la investigación sobre el Síndrome de Ehlers-Danlos se concentra, sobre todo, en Dinamarca y Suiza, pues es en estos países donde la enfermedad tiene mayor prevalencia. La mayoría de ellos forman parte de la Ehlers-Danlos Society, una comunidad global formada por pacientes, médicos e investigadores dedicada al soporte en investigación, así como a la divulgación de este síndrome y otros del espectro de la hipermovilidad.
¿Cómo se manifiesta?
Existe una gran variabilidad en las manifestaciones clínicas. Es una entidad clínica compleja integrada por un grupo de trastornos caracterizados por hiperextensibilidad de la piel, laxitud articular, fragilidad de la piel y de otros tejidos conectivos. Se conocen hasta 11 variantes distintas de Ehlers Danlos, cuya clasificación y nomenclatura están en continua revisión. Todas ellas comparten, generalmente:
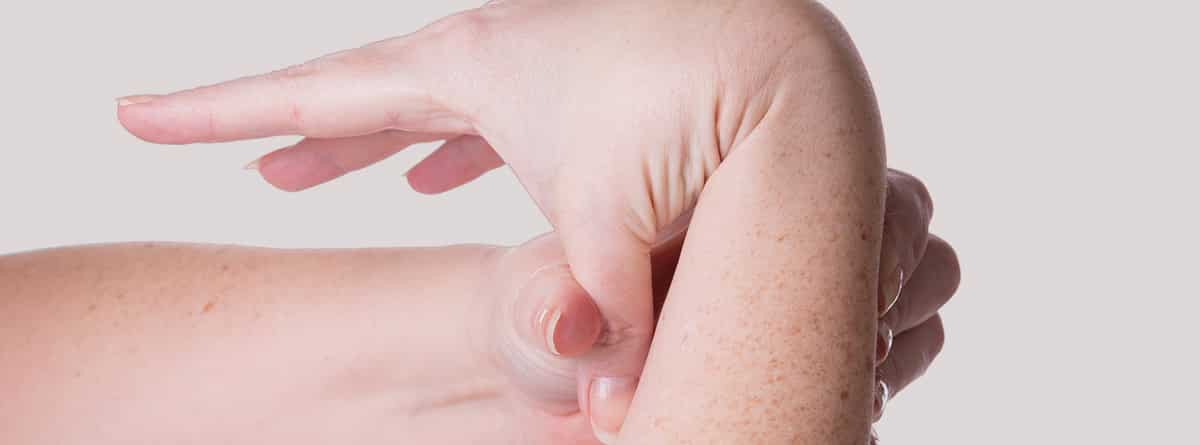
- Hiperlaxitud articular,
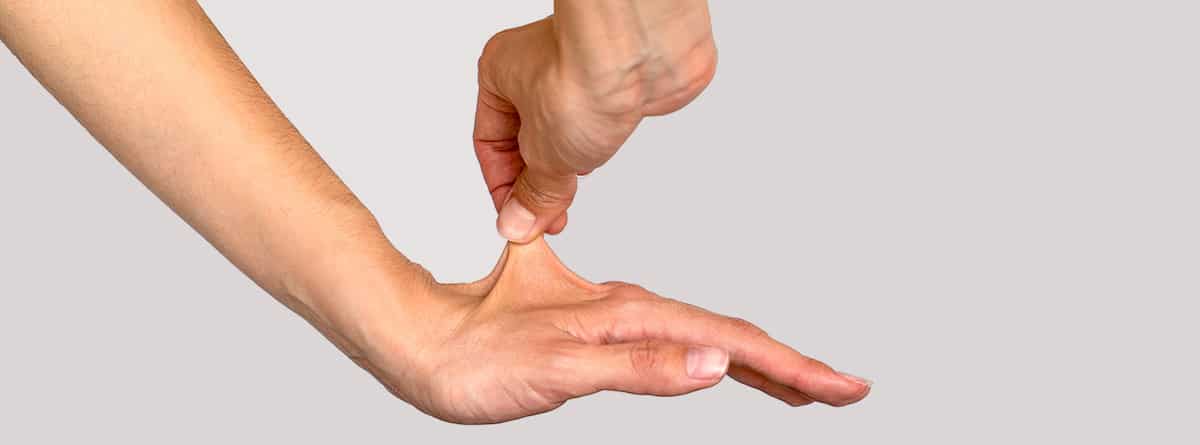
- Piel fina, frágil y elástica,
- Presencia de cicatrices en codos y rodillas secundarias a traumatismos,
- Debilidad muscular,
- Baja densidad ósea (u osteoporosis temprana)
- Escoliosis.
Por lo general, se verán afectados órganos con mucho tejido conjuntivo: la dermis, la aorta y carótida, las cavidades corporales, los ligamentos, los huesos, cartílago y el globo ocular.
Síntomas
- Los pacientes con este síndrome suelen tener dolor crónico en articulaciones y baja densidad ósea, derivado de problemas de la absorción del calcio. El dolor musculoesquelético de carácter crónico, severo, debilitante, refractario al tratamiento y afecta a varias articulaciones. El tratamiento a seguir es paliativo, se centra en las complicaciones que pueda generar cada tipo.
- La cara de estos pacientes tiene un aspecto característico, denominado facies acrogérica: nariz fina, labios delgados, mejillas delgadas y ojos prominentes.
- La piel de las manos y de los pies puede ser extremadamente fina con aspecto de envejecimiento prematuro (acrogeria).
Diagnóstico
Para el diagnóstico del síndrome de Ehlers Danlos no existen pruebas específicas. En muchos casos cuesta años llegar a encontrar que la causa de los problemas es esta entidad. Uno de los puntos más importantes es tener en cuenta la historia familiar, ya que es posible que haya antecedentes en la familia:
- Manifestaciones clínicas,
- Historia familiar compatible
- Demostración del defecto en el colágeno.
El diagnóstico diferencial es con otras enfermedades del tejido conectivo, como el síndrome de Marfan y la acrogeria de Gottron.
Criterios diagnósticos
Los criterios mayores o principales del síndrome de Ehrlers Danlos clásico son:
- Hiperextensibilidad cutánea y problemas de cicatrización.
- Hipermovilidad articular generalizada.
Además, presenta criterios menores como:
- Facilidad para hacerse hematomas.
- Piel suave y aterciopelada.
- Fragilidad cutánea.
- Lesiones subcutáneos.
- Hernias.
- Pliegues epicánticos.
- Complicaciones de la hipermovilidad articular: esguinces, subluxaciones, luxaciones, dolor o pies planos.
- Familiar de primer grado con este síndrome.
Posibles complicaciones
Complicaciones vasculares
- Hematomas,
- Aneurismas (bolsa formada por la dilatación o rotura de las paredes de una arteria o vena) arteriales,
- Falsos aneurismas,
- Fístulas (comunicación anormal entre dos órganos internos o hacia la superficie corporal) arterio-venosas,
- Disecciones y roturas de los vasos. Las rupturas arteriales son responsables de la mayoría de los fallecimientos pues son frecuentes, e imprevisibles y con una reparación quirúrgica muy difícil debido a la friabilidad de los tejidos. Son raros, aunque están descritos, los aneurismas disecantes de la aorta, la ruptura aórtica.
Complicaciones del sistema nervioso central
Pueden ser aneurismas disecantes de los vasos intracraneales y fístulas carótideas.
No existe predisposición familiar por un tipo especial de complicación, presentarse diferentes tipos de trastornos en diferentes miembros de una familia y en un mismo individuo.
Lo que debes saber…
- Es una entidad clínica compleja integrada por un grupo de trastornos caracterizados por hiperextensibilidad de la piel, laxitud articular, fragilidad de la piel y de otros tejidos conectivos.
- Este síndrome se refiere a un conjunto de enfermedades heterogéneas hereditarias relacionadas con la alteración genética tanto de la estructura como la síntesis del colágeno.
- El tratamiento a seguir es paliativo, se centra en las complicaciones que pueda generar cada tipo.
Recuerda que en MAPFRE cuidamos tu bienestar y el de los tuyos, por eso te ofrecemos un Seguro de Salud con las mejores coberturas para que siempre estés protegido ante cualquier patología o enfermedad.


Comentarios (0)